Chapter 5 RNAseq Analysis
RNAseq (RNA-sequencing) is a technique that can examine the quantity and sequences of RNA in a sample using next generation sequencing (NGS). It analyzes the transcriptome of gene expression patterns encoded within our RNA.
The following course provides an excellent overview of Next Generation Sequencing (NGS) and Illumina NGS Sample Preparation. Please take the time to watch the videos!
Next Generation Sequencing 1 - Overview
Next Generation Sequencing 2 - Sample Preparation
In this talk, Eric Chow explains the chemistry behind next generation sequencing, and describes how the next gen sequencers detect and display results. The most commonly used Illumina sequencers are image based and detect the addition of fluorescently labelled nucleotides. Chow also describes two different next generation sequencing technologies which provide benefits such as much longer reads but with downsides such as higher error rates. Chow finishes the talk with some insights into medical applications of next gen sequencing such as much less invasive prenatal testing or cancer detection.
In his second talk, Chow discusses Illumina NGS Sample Preparation. He goes over DNA and RNA preparation, bead-based (Ampure or SPRI) cleanups, and sample quantification and quality control.
— https://www.ibiology.org/techniques/next-generation-sequencing/
5.1 RNAseq downstream analysis
5.1.1 nf-core/rnaseq pipeline - installation
![]()
nf-core/rnaseq is a bioinformatics analysis pipeline used for RNA sequencing data. The pipeline is built using Nextflow, a workflow tool to run tasks across multiple compute infrastructures in a portable manner. Detailed documentation on how to install and run nextflow and nf-core bioinformatic pipelines can be found at the following sites:
For your convenience install instructions are reproduced below.
5.1.2 Nextflow
All nf-core pipelines use Nextflow, so this must be present on the system where you launch your analysis. See nextflow.io for the latest installation instructions.
Nextflow runs on most POSIX systems (Linux, Mac OSX etc) and can typically be installed by running the following commands:
# Make sure that Java v8+ is installed:
java -version
# Install Nextflow
curl -fsSL get.nextflow.io | bash
# Add Nextflow binary to your user's PATH:
mv nextflow ~/bin/
# OR system-wide installation:
# sudo mv nextflow /usr/local/binYou can also install Nextflow using Bioconda:
conda install -c bioconda nextflow5.1.3 Additional pipeline software
nf-core pipelines utilise Docker, Singularity and Conda to seamlessly install and run different software packages. These packages create isolated compute environments on the fly allowing for flexibility in development and delivery.
- Docker | Typically used locally / on single-user servers and the cloud. Analysis runs in a container, which behaves like an isolated operating system Previously required system root access, though a “rootless mode” is available since late 2019
- Singularity | Often used as an alternative to Docker on multi-user systems such as HPC systems. Also runs containers and can create these from Docker images Does not need root access or any daemon processes - images built from files
- Conda | Packaging system that manages environments instead of running analysis in containers. Poorer reproducibility than Docker / Singularity There can be changes in low-level package dependencies over time The software still runs in your native operating system environment and so core system functions can differ
5.1.4 Pipeline code
The nf-core pipeline does not need to be installed. On calling nextflow nf-core/rnaseq ..., pipeline components are automatically downloaded from github.
5.1.5 Reference genomes
Some pipelines come with built-in support for iGenomes references. It may be preferable for you to download a local copy of these to your system so as to avoid fetching the same reference many times. For more information, see Reference genomes.
5.2 Running the nf-core/rnaseq pipeline
Running the nf-core/rnaseq pipeline is incredibly simple and requires only a basic setup.
5.2.1 Setup
The nf-core/rnaseq pipeline typically requires a sample sheet as input. The sample sheet should comprise the following columns.
| Column | Description |
|---|---|
| group | Group identifier for sample. This will be identical for replicate samples from the same experimental group. |
| replicate | Integer representing replicate number. Must start from 1.. |
| fastq_1 | Full path to FastQ file for read 1. File has to be zipped and have the extension “.fastq.gz” or “.fq.gz.” |
| fastq_2 | Full path to FastQ file for read 2. File has to be zipped and have the extension “.fastq.gz” or “.fq.gz.” |
| strandedness | Sample strand-specificity. Must be one of unstranded, forward or reverse. |
A final design file consisting of paired-end data may look something like the one below - see Table 3.1. This sample sheet represents a full sized RNAseq dataset obtained from the ENCODE project, which is routinely used to test the nf-core/rnaseq pipeline and is described here
sampleSheet <- read.delim("RNASeqData/samplesheet.valid.csv",
stringsAsFactors = FALSE, sep = ",")
head(sampleSheet)
#> sample single_end
#> 1 GM12878_R1_T1 0
#> 2 GM12878_R2_T1 0
#> 3 H1_R1_T1 0
#> 4 H1_R2_T1 0
#> 5 K562_R1_T1 0
#> 6 K562_R2_T1 0
#> fastq_1
#> 1 s3://nf-core-awsmegatests/rnaseq/input_data/SRX1603629_T1_1.fastq.gz
#> 2 s3://nf-core-awsmegatests/rnaseq/input_data/SRX1603630_T1_1.fastq.gz
#> 3 s3://nf-core-awsmegatests/rnaseq/input_data/SRX2370468_T1_1.fastq.gz
#> 4 s3://nf-core-awsmegatests/rnaseq/input_data/SRX2370469_T1_1.fastq.gz
#> 5 s3://nf-core-awsmegatests/rnaseq/input_data/SRX1603392_T1_1.fastq.gz
#> 6 s3://nf-core-awsmegatests/rnaseq/input_data/SRX1603393_T1_1.fastq.gz
#> fastq_2
#> 1 s3://nf-core-awsmegatests/rnaseq/input_data/SRX1603629_T1_2.fastq.gz
#> 2 s3://nf-core-awsmegatests/rnaseq/input_data/SRX1603630_T1_2.fastq.gz
#> 3 s3://nf-core-awsmegatests/rnaseq/input_data/SRX2370468_T1_2.fastq.gz
#> 4 s3://nf-core-awsmegatests/rnaseq/input_data/SRX2370469_T1_2.fastq.gz
#> 5 s3://nf-core-awsmegatests/rnaseq/input_data/SRX1603392_T1_2.fastq.gz
#> 6 s3://nf-core-awsmegatests/rnaseq/input_data/SRX1603393_T1_2.fastq.gz
#> strandedness
#> 1 reverse
#> 2 reverse
#> 3 reverse
#> 4 reverse
#> 5 reverse
#> 6 reverse5.3 Running the nf-core/rnaseq pipeline
Once the setup is complete, running the nf-core/rnaseq pipeline is as simple as typing the following into the command line:
nextflow run nf-core/rnaseq \
--input samplesheet.csv \
--fasta '<PATH TO FASTA FILE>/genome.fa'\
--gtf '<PATH TO FASTA FILE>/genome.gtf'
--aligner star_rsem
-profile conda5.4 Quality Control (QC) and Merged count files
After run completion, the nf-core pipeline will generate a number of data files that can be used to assess sequence quality and for downstream analysis. A typical run folder will comprise the following folder structure. Please see Run Folder Example.
5.4.1 QC Metrics
The first step after the completion of any analysis run is to check the QC metrics. A summary of all QC metrics for a given run can be found in the file named multiqc_report.html, which is located in the MultiQC folder. A detailed overview of the content provided in the MultiQC report can be found at the following location https://nf-co.re/rnaseq/3.0/output
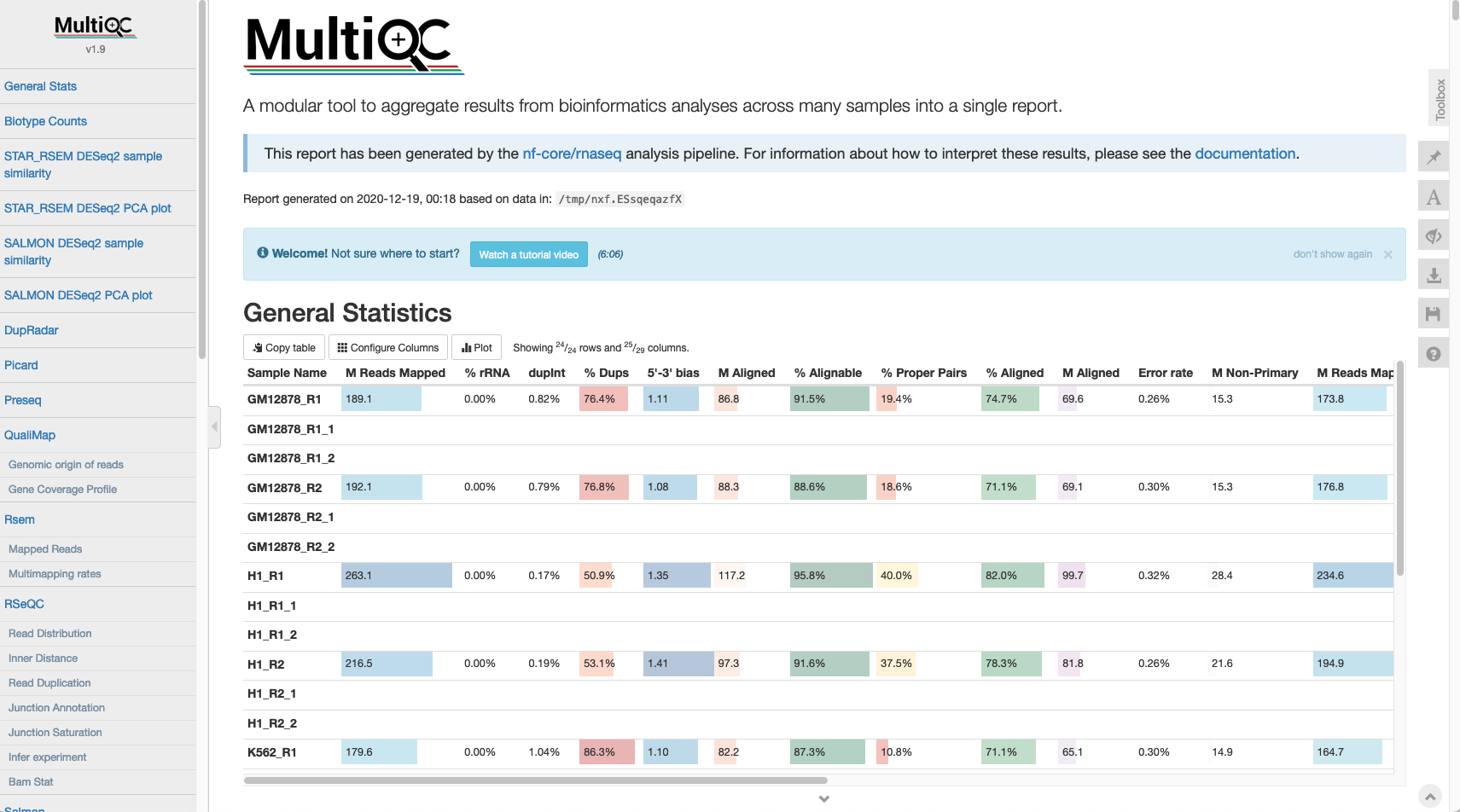
5.4.2 Merged count files
After assessing QC, we are ready to start a primary analysis of the data. To do so we will typically use a merged gene count file. For example, rsem.merged.gene_counts.tsv. These files typically comprise the Ensembl Gene ID, Ensembl Transcript ID and Sample IDs with each column/row entry representing the read count for a given gene. See example below:
mergedCounts <- read.delim("RNASeqData/rsem.merged.gene_counts.tsv",
stringsAsFactors = FALSE, sep = "\t")
colnames(mergedCounts)
#> [1] "gene_id" "transcript_id.s." "GM12878_R1"
#> [4] "GM12878_R2" "H1_R1" "H1_R2"
#> [7] "K562_R1" "K562_R2" "MCF7_R1"
#> [10] "MCF7_R2"
mergedCounts[1:5,c(1,3:7)]
#> gene_id GM12878_R1 GM12878_R2 H1_R1 H1_R2 K562_R1
#> 1 ENSG00000000003 7 0 8299 4390 3
#> 2 ENSG00000000005 0 0 69 30 0
#> 3 ENSG00000000419 4061 3582 1885 796 6194
#> 4 ENSG00000000457 1942 1606 592 409 918
#> 5 ENSG00000000460 1862 1612 3594 1748 2813